Molecular dynamics to fight infection
Department of biochemistry, microbiology and bio-informatic, University Laval
Mode of action of the protein truncated hemoglobin N in the infection process of tuberculosis bacteria

Patrick Lagüe, biophysicist
Tuberculosis infects one-third of the world’s population. In the majority of the people infected, the immune system succeed to stop the progression of the disease by producing NO and reducing the concentration of O2 in the infected lesion. Under those conditions, the NO produces a toxic effect that affects the respiration of the bacteria and maintains them in a latent state that has no consequences for the organism. However, the tuberculosis bacteria have a defense system that fights the poisoning.  A protein (truncated hemoglobin N, trHbN) can transform the NO into nitrate and therefore protect their respiration. Because of its role in the survival of the tuberculosis bacteria, the characterization of the structure and function of the trHbN has become a point of interest. To do so, we use models of molecular dynamics (MD) that consist in simulation of movement from a group of atoms (6000) in time under known conditions. A dynamic algorithm is used to calculate forces as well as speed and positions of the atoms that inform on the molecular trajectory during a specific time (17.9 µs). These computations use 150 core-year on the supercomputer called Colossus. These simulations allowed to identified the specific aspect of the structure of this protein that gives rise to this high reactivity to NO. Cavities are found in the highly rigid backbone of the protein. The hydrophobic nature of the cavities force water molecules to partition away from the protein surface and thus favor NO capture. To better understand the environmental impact on the structure and function of the protein, molecular dynamic simulation were also used to study the protein in milieu of different compositions. The magnitude of the interaction of the protein with the biological membrane depends on the nature of the lipid composing the membrane. The interaction is greater with the lipid cardiolipin that is found almost exclusively in the membranes that build the walls of bacteria. Such environment can bring advantages to the enzymatic activity of the protein.
A protein (truncated hemoglobin N, trHbN) can transform the NO into nitrate and therefore protect their respiration. Because of its role in the survival of the tuberculosis bacteria, the characterization of the structure and function of the trHbN has become a point of interest. To do so, we use models of molecular dynamics (MD) that consist in simulation of movement from a group of atoms (6000) in time under known conditions. A dynamic algorithm is used to calculate forces as well as speed and positions of the atoms that inform on the molecular trajectory during a specific time (17.9 µs). These computations use 150 core-year on the supercomputer called Colossus. These simulations allowed to identified the specific aspect of the structure of this protein that gives rise to this high reactivity to NO. Cavities are found in the highly rigid backbone of the protein. The hydrophobic nature of the cavities force water molecules to partition away from the protein surface and thus favor NO capture. To better understand the environmental impact on the structure and function of the protein, molecular dynamic simulation were also used to study the protein in milieu of different compositions. The magnitude of the interaction of the protein with the biological membrane depends on the nature of the lipid composing the membrane. The interaction is greater with the lipid cardiolipin that is found almost exclusively in the membranes that build the walls of bacteria. Such environment can bring advantages to the enzymatic activity of the protein.
Mode of action of the fusion peptide in the infection process of influenza virus
During infection with influenza virus, the virus penetrates into cells then it fusions with the membrane of the endosome. 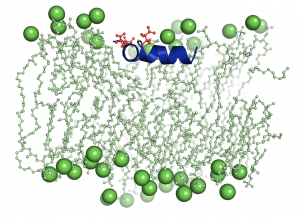 The endosome is a compartment in the cell that sorts proteins, recycling those in its membrane and degrading the rest. The low PH in the endosome induce the insertion of the viral protein hemagglutinin in the membrane and hence its recycling. More specifically, it is the subunit of the protein called fusion peptide that gets in contact with the membrane. The understanding of this phenomenon is essential to develop therapy to stop infection. MD simulations of a group of 30000 atoms for 10µs are used to study the behavior of the molecular structure of the fusion peptide in face of lipids membrane and depending on PH. Those computations need 44 cores-year from Colosse.
The endosome is a compartment in the cell that sorts proteins, recycling those in its membrane and degrading the rest. The low PH in the endosome induce the insertion of the viral protein hemagglutinin in the membrane and hence its recycling. More specifically, it is the subunit of the protein called fusion peptide that gets in contact with the membrane. The understanding of this phenomenon is essential to develop therapy to stop infection. MD simulations of a group of 30000 atoms for 10µs are used to study the behavior of the molecular structure of the fusion peptide in face of lipids membrane and depending on PH. Those computations need 44 cores-year from Colosse.
Rational design of new antibiotics
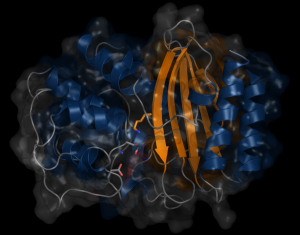
β-lactamases dynamic properties
β-lactams is the most used antibiotics class on the market. β-Lactamases are enzymes found in bacteria that break the β-lactams and thus confer antibiotics resistance to bacteria. TEM-1 confers ampicillin resistance to E. coli and PSE-4 confers carbenicillin resistance to P. aeruginosa. For new antibiotic engineering, it is important to acquire more information on the mechanism that confers resistance to bacteria. We need to understand the molecular motions of TEM-1 and PSE-4 that allows the antibiotics recognition and rupture. The properties of these proteins dynamics will be characterized with the help of MD combined to RMN spectroscopy that measures the magnetic interactions between molecules.
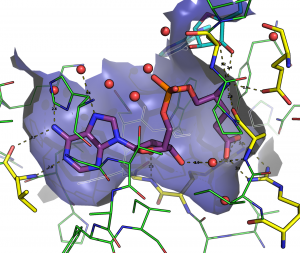
The Glutamyl-tRNA synthétase as therapeutic target
Glutamyl-tRNA synthetase is an essential protein which inhibition kills the bacteria. The goal being to develop a new class of antibiotics, we study the inhibitors of the GluRS and more specifically the interactions between the molecules of the protein and 50 different ligands. To characterize these interactions, we use MD simulation of 60000 atoms during 25 µs for a total of 100 cores-year on Colossus. Two more techniques are used, the docking (fitted) and quantum mechanics (GAMESS).
